- Visibility 114 Views
- Downloads 9 Downloads
- DOI 10.18231/j.ijpca.2024.012
-
CrossMark

Design, formulation and in-vitro evaluation of diclofenac sodium fast disintegrating tablets
- Author Details:
-
 P SS Prasanna Kumar *
P SS Prasanna Kumar *
-
T Anjali
-
Srinivas Nandyala
-
K. Sivaji
Introduction
Drug delivery is the most important and vastly using technique for administration of an API to show pharmacological activity, achieve a therapeutic effect, primarily due to patient acceptability, ease of administration, inexpensive manufacturing process and release pattern. For many drugs, tablets incorporated with rapid disintegrating agent provide clinically effective treatment thereby maintains the required balance of pharmacokinetic and pharmacodynamic profiles of API with acceptable levels of patient safety and efficacy. These API and excipients are designed to formulate and to provide maximum stability, activity and bioavailability.[1]
Fast disintegrating tablets are those that disintegrate rapidly and release the drug by dissolving. A disintegrant is belongs to the tablet excipient, and it was incoeporated to dosage form which we have chosen either it maybe a tablet or capsule for breakup of the compacted mass when it is enter into a fluid environment(gastric fluid or intestine).[2] Disintegrants are added to tablets or encapsulated formulations to break the tablet or capsule slugs into smaller fragments in the presence of solvent(aqueous) environment, thereby increasing the available particle surface area promote API to release more rapidly.[3], [4]
Materials and Methods
Isolation of corn silk powder: The collected Fresh corn silk cleaned with tap water, soaked in a distilled water, dried in oven at 45oC upto constant weight. The obtained dried silk was converted into a state of fine powder and it was extracted with 40% v/v ethanol - distilled water. The dried sample macerated with three times of 40% v/v ethanol (1.5 L) for 24 h at room temperature,digested with distilled water (1.5 L) for 1 h at 45oC. The collected extract solution was separately filtered using filter paper and the used solvents evaporated at 40oC. The final sample was dried and used in the form of solid powder and stored at 20oC.[5]

Preformulation study
Organoleptic Characteristics:
Colour: A small amount of API was viewed in well illuminated place
Odour,Taste: Odour and taste were flavoured using tongue and smell
Determination of melting point: Take small portion of drug containing capillary tube which is closed at one end and placed in melting point apparatus, the temperature at which melting of drug occur was observed. Perform the test for 3 times and note the average of 3 values to get accuracy.[6]
Solubility Analysis: A supersaturated solution of medicament was prepared using water and also with 0.1N HCl, Selected Phosphate buffers-pH 6.4, 6.8,7.2 separately. The aliquots prepared were kept aside for 24hrs and followed by filtering. Collect the filtrate, measure the absorbance at 273nm by UV spectrophotometer.
The solubility can be done by
Solubility = (At/As) ×Cs× (Dt/Wd×1000) ×100
Where At = Absorbance of Sample
As -=Absorbance of Standard absorbance
Cs-=Standard concentration of drug.
Dt = Dilution factor.
Wd-=Weight of the drug.
Pre-Compression Evaluation Parameters
Angle of repose: To evaluate flow property of powder blend by Fixed funnel method. Weigh the specified quantity of blend taken in a funnel, height of funnel was adjusted. The tip of funnel just touches the apex of powder heap. The powder flow freely allowed through a funnel to the surface. The diameter, height and radius of powder cone was measured and calculate by incorporating the obtained values.
θ =tan-1(h/r)
Where θ =angle of repose
h =height of cone in cm
r =radius of the cone base in cm.
Bulk density (ρb): The bulk density was calculated byBulk density (ρb) = Mass of the powder (M) / Bulk volume of powder (Vb) Tapped density (ρt): It was calculated by using the following formula.Tapped density (ρt) = Mass of powder (M) / Tapped volume of powder (Vb)
Compressibility index or Carr’s index: To determine the flow property for precompression powder blend using tapped density and bulk density.
% of Carr’s index = ρt – ρb / ρt ×100Where ρt = tapped density of powderρb = bulk density of powder.Hausner’s ratio: The ratio of tapped density to bulk density.Hausner’s ratio = Vi / VoWhere, Vo = Bulk density Vi = Tapped density
Drug Excipient Compatibility Studies
Fourier Transform Infrared Spectroscopy (FTIR): The objective involved in FT-IR studies between drug and excipient compatibility considerations. The samples of homogenous mixtures prepared with Drug and excipients, filled in glass vials subjected to accelerated stability studies, maintained at 60± 2°C for 02 weeks.
The FTIR spectrum of pure Diclofenac sodium is compared with spectrum obtained from mixture of Diclofenac sodium along with the selected excipients. Any changes in peak intensities, shifts, or the appearance/disappearance of peaks, indicating potential interactions between drug, excipients in the spectra were interpreted.
Analytical method: Calibration curve of Diclofenac sodium
Stock I solution: 25mg of Diclofenac sodium in 25ml of phosphate buffer of PH 6.8 solution(1mg/1ml).
Stock II solution: Transfer 10ml stock I solution to a 100ml volumetric flask, dilute with Phosphate buffer PH 6.8 (100µg/1ml).
Sample Preparation: From stock II solutions number of concentrations from 0-10 μg/mL are diluted using Phosphate buffer PH 6.8, measure the absorbance using UV double beam spectrophotometer at 273nm.[6], [7], [8], [9], [10], [11], [12], [13], [14]
Formulation and development of Diclofenac sodium fast disintegrating tablet (Direct Compression method)
Preparation of Ingredients- Diclofenac sodium, Disintegrating agent, corn silk powder and lactose individually weighed.
Blending-The ingredients are sifted through a sieve no. 40 followed by blending in a poly bag for 10 min.
Lubrication-Add Weighed quantity of Magnesium Stearate, sieved through a sieve no. 60 to the above prepared blend.
Compression-The blend lubricated is compressed using Upper and lower punch.
|
S. No |
Name of the ingredients |
F1 |
F2 |
F3 |
F4 |
F5 |
|
1. |
Diclofenac sodium |
100 |
100 |
100 |
100 |
100 |
|
2. |
Corn silk powder |
04 |
08 |
12 |
16 |
20 |
|
3. |
Magnesium stearate |
03 |
03 |
03 |
03 |
03 |
|
4. |
Talc |
03 |
03 |
03 |
03 |
03 |
|
5. |
Lactose |
90 |
86 |
82 |
78 |
74 |
|
|
Total weight in mg |
200 |
200 |
200 |
200 |
200 |
Post-compression Evaluation tests: [6], [7], [8], [9], [10], [11], [12], [13], [14]
General appearance: The observations for shape, colour, texture and odour of formulated tablets.Thickness: Vernier Calipers is used to measure thickness of the tablets.Hardness: Force required to break a tablet under applied pressure using Pfizer/ Monsanto hardness tester. Position the tablet between two anvils of apparatus. Apply pressure and measure the crushing strength which leads the tablet to break and force exerted on the tablet fracture is recorded. Therefore, hardness is called Crushing Strength. By measuring tablet hardness, pharmaceutical manufacturers can ensure that their products meet the required standards for mechanical strength, which is essential for maintaining product integrity and performance throughout the manufacturing process and during storage and transportation.Friability: The test helps in ensuring tablet durability and integrity so it is defined as loss in weight of tablet in the container due to removal of fine particle from their surface and expressed in percentage (%).
% Friability calculated by using the following formula:
Friability= [(Initial weight of tablets – Final weight of tablets)/ Initial weight of tablets] X 100
Weight Variation: It test helps in knowing the content uniformity and consistency of the final product.
Weighed Individually 20 tablets, to find out the average weight not more than two of the individual weights deviate from the average weight by more than the percentage deviation.
Disintegration Test: The ability of tablets to rapidly disintegrate into smaller particles when exposed to disintegration fluid under controlled conditions. The in-vitro disintegration time determined by using disintegration test apparatus. Place one tablet in each of the 6 tubes of the basket, add a disc to each tube and run the apparatus with disintegration fluid like water,0.1 N HCl or Phosphate buffer PH 6.8. Ensure that to maintain the temperature at 37°C ± 2°C. Observe and record time taken for the tablets to disintegrate.
Content uniformity (Assay): The test ensures that each tablet in batch contains specified amount of active ingredient. Weigh and powder 20 tablets equivalent to 50mg of drug and shake with 60ml of methanol in 200ml of volumetric flask. Dilute the volume using methanol, sonicated for 30 minutes. The aliquot of 5ml dilute with methanol up to 100ml and Centrifuge at 10,000 RPM up to 10 min. By using nylon membrane (0.45µ) to Filter, measure the absorbance at 273nm.
Assay = (At/As)×Cs× (Dt/Wd) ×100
Wetting Time: This method is utilized to assess the wetting properties of tablets which plays vital role in drug delivery and formulation purposes. The time for complete wetting of the tablet. A piece of tissue paper folded twice placed in a small Petri dish with 10ml of water, a water-soluble dye and the tablet was placed on this paper. The conclusion about wetting the tablets based on observed wetting time.
In-Vitro drug release: The study of this parameter is very crucial for assessing the performance and effectiveness of the formulations. The release of rate of tablets was determined using USP dissolution testing apparatus type II (Paddle method). The dissolution was performed with 900 ml of Phosphate buffer PH 6.8, at 370± 0.50 C and 50 rpm. At specified time interval 5ml of dissolution medium samples were withdrawn from the dissolution apparatus and replacement of fresh fluid and filtered through 0.45μm and measure the absorbance at 273nm using UV /Visible Spectrophotometer The drug release was plotted against time. This graph obtained provide insights into the dissolution behavior of the fast-disintegrating tablets over time. The % drug release of the can be calculated by
% Drug release = (At/As) ×Cs× (Dt ×Vm/Wd×1000) ×100
Where At =Sample absorbance.
In-Vitro Release Kinetics Studies: [6], [7], [8], [9], [10], [11], [12], [13], [14] Based on the analysis, the mechanism of drug release zero-order or first-order kinetics of order can be determined. This information is crucial for understanding the drug release profile of dosage form, optimizing formulation parameters, and predicting in- vivo behavior.
Zero Order Kinetics: A linear relationship between the fractions of drug released with time.
Q=ko t.
Where, Q = fraction of drug released at time t & Ko = zero-order release rate constant.
A plot of the % of drug released against time (Min) will be linear indicates zero order release kinetics.
First order kinetics: Wagner hypothesis suggests that exposed surface area of a tablet decreased exponentially with time during dissolution process suggested that the drug release from most of the slow-release tablets could be described adequately by the first-order kinetics. The equation for first order kinetics is
Log C= Log C0-kt/2.303
Results & Discussion
|
S. No |
Characteristics |
Results |
|
1 |
Description |
White |
|
2 |
Melting Point |
286oC±0.35 |
|
3 |
Bulk Density |
0.394±0.46 g/ml |
|
4 |
Tapped Density |
0.443±0.06 g/ml |
|
5 |
Carr’s Index |
11.96±0.12% |
|
6 |
Hausner’s Ratio |
1.12±0.06 |
|
7 |
Angle of Repose |
28.350±0.127 |
Discussion: From the observed results, it was concluded that API exhibited good flow property.
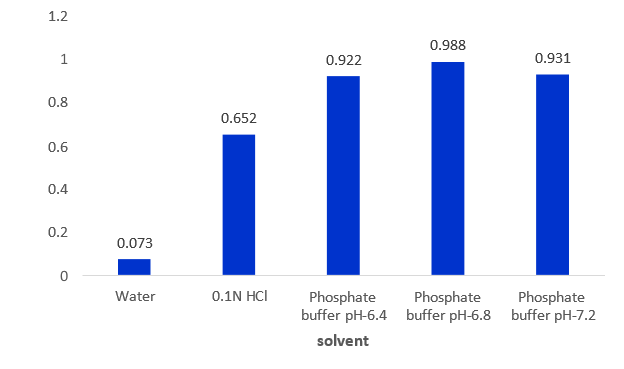
Discussion: The solubility of Diclofenac sodium was determined by using different solvents from that it was observed the maximum solubility was showed with Phosphate buffer pH-6.8, minimum solubility in water.
|
S. No |
Characteristics |
Corn silk |
|
1 |
Description |
Pale yellow |
|
2 |
Bulk Density |
0.29±1.12gm/ml |
|
3 |
Tapped Density |
0.33±0.83gm/ml |
|
4 |
Carr’s Index |
12.12% |
|
5 |
Hausner’s Ratio |
1.13gm/ml |
|
6 |
Angle of Repose |
33.02±1.42 |
Discussion: From the above results, it was concluded that the selected Natural polymers Corn silk powder exhibited good flow property.
Standard Calibration Curve of Diclofenac sodium-The λ max was obtained at 273 nm with Phosphate buffer at pH 6.8. Th regression value is 0.999 for for the concentration range of 0 to 10 µg/mL. The calculation of In-vitro drug release and Assay was based on this calibration curve.
|
S. No |
Concentration (μg/ml) |
Absorbance at 273nm |
|
1. |
0 |
0 |
|
2. |
02 |
0.144±0.020 |
|
3. |
04 |
0.279±0.006 |
|
4. |
06 |
0.422±0.018 |
|
5. |
08 |
0.554±0.023 |
|
6. |
10 |
0.690±0.019 |
|
Formulation Code |
Angle of repose (Ɵ) |
Bulk Density (gm/cc) |
Tapped Density (gm/cc) |
Compressibility (%) |
Hausner’s ratio |
|
F1 |
32.96±0.13 |
0.578±0.11 |
0.656±0.35 |
11.89±0.32 |
1.13±0.23 |
|
F2 |
34.73±0.52 |
0.746±0.55 |
0.862±0.52 |
13.4±0.43 |
1.15±0.36 |
|
F3 |
31.19±0.15 |
0543±0.10 |
0.621±0.63 |
12.56±0.48 |
1.14±0.74 |
|
F4 |
32.47±0.42 |
0.624±0.25 |
0.713±0.42 |
12.48±0.42 |
1.14±0.21 |
|
F5 |
34.19±0.25 |
0.644±0.32 |
0.730±0.75 |
11.76±0.24 |
1.13±0.16 |
|
Formulations |
Weight variation (mg) |
Thickness (mm) |
Hardness (kg/cm2) |
Friability (%) |
Assay (%) |
Disintegrat ion time(sec) |
Wetting time(sec) |
|
F1 |
200 |
2.80±0.87 |
4.48±0.02 |
0.64±0.28 |
98.03±0.27 |
85±0.68 |
56±0.47 |
|
F2 |
199.2 |
2.79±0.22 |
4.61±0.41 |
0.62±0.54 |
99.53±0.43 |
70±0.52 |
43±0.43 |
|
F3 |
199.01 |
2.72±0.49 |
4.26±0.04 |
0.68±0.31 |
100.06±0.36 |
67±0.47 |
35±0.21 |
|
F4 |
199.5 |
2.96±0.36 |
4.65±0.25 |
0.73±0.04 |
99.13±0.09 |
52±0.43 |
29±0.16 |
|
F5 |
199.3 |
2.99±0.28 |
4.52±0.32 |
0.56±0.35 |
100.02±0.17 |
28±0.12 |
20±0.09 |
|
Time (Min) |
F1 |
F2 |
F3 |
F4 |
F5 |
|
0 |
0 |
0 |
0 |
0 |
0 |
|
5 |
11.29±0.05 |
16.44±0.21 |
17.97±0.11 |
24.33±0.23 |
21.33±0.23 |
|
10 |
26.55±0.09 |
30.02±0.15 |
33.32±0.05 |
36.21±0.17 |
38.21±0.17 |
|
15 |
40.16±0.13 |
42.73±0.35 |
55.95±0.21 |
51.33±0.03 |
55.33±0.03 |
|
20 |
54.72±0.11 |
58.8±0.11 |
72.08±0.23 |
75.10±0.15 |
79.10±0.15 |
|
25 |
64.52±0.23 |
72.08±0.09 |
86.36±0.41 |
91.23±0.24 |
99.98±0.06 |
|
30 |
81.93±0.05 |
81.43±0.21 |
92.44±0.04 |
99.92±0.04 |
|
|
40 |
92.03±0.21 |
86.56±0.08 |
99.88±0.09 |
|
|
|
50 |
94.32±0.45 |
99.61±0.05 |
|
|
|
|
60 |
99.62±0.11 |
|
|
|
|
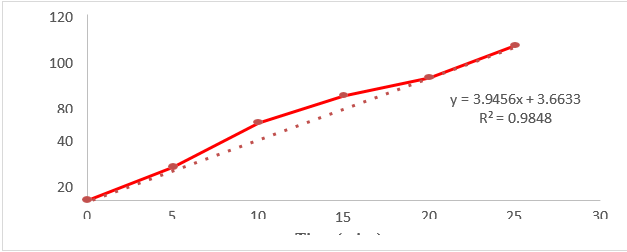
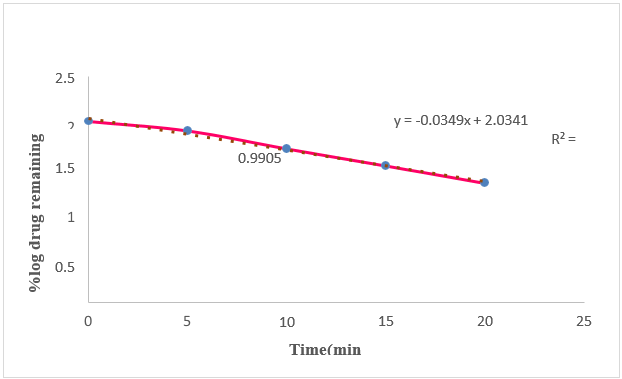
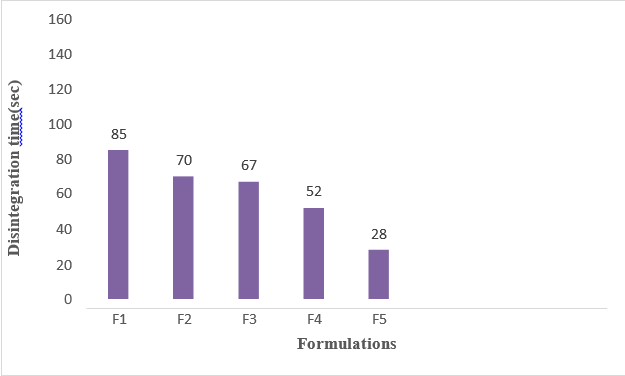
Among the formulations selected Natural Polymer corn silk acts as a Fast Disintegrating agents based on the concentration value increased then the disintegration time was decreased, increased dissolution rate which can be beneficial for improving drug absorption, onset of action.
Summary and Conclusion
The study was aimed to develop formulate in-vitro evaluation of Diclofenac sodium with corn silk powder with varying concentrations. Various physical characteristic tests were performed for powder blend which were found to be in acceptable limits. And also performed post compression tests for 05 the formulations the obtained results were found to be acceptable limit. Among all the 5 formulations, In-Vitro dissolution studies showed 99.98 % of drug release within 25 minutes for "F5" formulation showed better drug release compared to other formulations due to the ratio of polymer increased then the Disintegration time was decreased and mechanism of drug release from the FDT release was followed First order kinetics.
Source of Funding
None.
Conflict of Interest
None.
References
- KPR Chowdary, S Kalyani. Recent Research On Matrix Tablets For Controlled Release-Int. Res J Pharm App Sci 2013. [Google Scholar]
- T Rhodes. . Modern Pharmaceutics 1990. [Google Scholar]
- V Ravichandiran. Modern Pharmaceutics. Rev J Pharm Res 2011. [Google Scholar]
- MP Wagh. Formulation and evaluation of fast dispersible tablets of aceclofenac using different superdisintegrant. Int J Pharm Pharm Sci 2010. [Google Scholar]
- C Limmatvapirat. Phytochemical analysis of baby corn silk extracts. J Ayur Integrat Med 2019. [Google Scholar]
- GNK Ganesh, M Shanmukhasrinivas, R Sureshkumar, N Jawahar, V Senthil, D Nagasamyvenkatesh. Preparation and Evaluation of Sustained Release MatrixTablet of Diclofenac Sodium using Natural Polymer. J Pharm Sci Res 2010. [Google Scholar]
- L Lachman, HA Lieberman, L Joseph. The theory and practice of Industrial pharmacy. 1990. [Google Scholar]
- . . Remington, the Science and practice of pharmacy 2002. [Google Scholar]
- N Alford, PJ Martin. . Martin’s Physical pharmacy and pharmaceutical sciences 2006. [Google Scholar]
- EN Madhu, R Karunakar, KK Pavan, GC Raghunadha. Development and evaluation of extended release matrix tablets of Alfuzosin HCl and its comparison with marketed product. J Pharm Res 2011. [Google Scholar]
- A Martin, J Swarbrick. . Physical pharmacy 1993. [Google Scholar]
- H Lieberman, L Lachman. Pharmaceutical dosage forms Tablets. . [Google Scholar]
- S Jain, SK Yadav, UK Patil. Preparation and evaluation of sustained release matrix tablet of furosemide using natural gum. Res J Pharm tech 2008. [Google Scholar]
- DM Treatise. . Biopharmaceutics and Pharmacokinetics -A Treatise 2009. [Google Scholar]
- Introduction
- Materials and Methods
- Pre-Compression Evaluation Parameters
- Drug Excipient Compatibility Studies
- Analytical method: Calibration curve of Diclofenac sodium
- Formulation and development of Diclofenac sodium fast disintegrating tablet (Direct Compression method)
- Results & Discussion
- Summary and Conclusion
- Source of Funding
- Conflict of Interest