Introduction
Eye cancer is a rare condition. The muscles, skin, and nerves that make up the eyelid and other external components of the eye may be impacted. If the cancer originates within the eyeball, it's termed intraocular cancer. The most prevalent intraocular tumors in adults are melanoma and lymphoma. The most prevalent eye cancer in children is retinoblastoma, which begins in the cells of the retina. Additionally, cancer can travel from other body parts to the eye. The most prevalent intraocular neoplasm in children, retinoblastoma is believed to affect 1 in 15,000 to 1 in 18,000 live births. It contributes to approximately four percent of cancers that occur in youngsters. In the United States, roughly 250 and 300 children acquire an outbreak of retinoblastoma year; considerably more cases are seen in developing nations. Statistics from India indicates a two to three times greater frequency of retinal tumors, with retinoblastoma being responsible for the bulk of cases in children around the age of fifteen. Likewise, only 50% of youngsters with retinoblastoma survive all over the world, compared to over 95% in the commercialized world. This could have been attributed to a longer time frame for presentation and, as an outcome, the greater frequency of extraocular invasion reported in developing countries. In developed countries, extraocular retinoblastoma is incredibly rare (reported incidence: around 2-5%). Fifty percent of retinoblastoma cases that present to a tertiary care referral center in developing countries have been triggered by extraocular disease. This high rate of advanced disease in developing countries can be explained by a few variables such as poverty, illiteracy, alternative health care systems, and restricted access to healthcare resources. We are going to explore the state of the art in retinoblastoma management throughout this article. We executed a PubMed search to discover important studies regarding the last 20 years of advancement in the management of retinoblastoma. We analyzed these articles, focusing specific consideration to the facts regarding retinoblastoma in India.1, 2, 3, 4
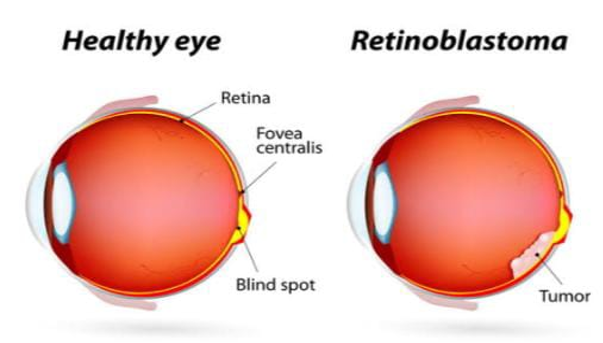
Genetics of Retinoblastoma
A mutation on chromosome 13, band 14 (13q14), long arm, is the root cause of it. The disease has been triggered by a mutation in the RB1 gene, a gene that inhibits tumor growth. A nongerminal disease which is not heritable appears in the child when both mutations only impact retinal cells, or somatic cells. On the other hand, the child contracts the germinal type of disease, which is genetic to the following generation, when an isolated mutation impacts the germinal cell. Of newly diagnosed cases of retinoblastoma, 94% are rare and only 6% are familial. Both retinoblastomas include germinal mutations in every scenario. whereas 85% of cases of unilateral sporadic retinoblastoma are occasional, 15% are due to germinal mutations affecting just one of the eyes.4
Risk for youngsters born to retinoblastoma survivors. Children of survivors of bilateral (hereditary) disease have a 45% risk of developing retinoblastoma, while those of survivors of unilateral retinoblastoma have a 2.5% risk.5
Siblings of retinoblastoma patients are at risk. Family members of patients with bilateral involvement have a 45% risk of developing retinoblastoma when the family background is positive, while siblings of patients with unilateral involvement have a 30% risk. Siblings of patients with bilateral disease have a 2% risk, whereas siblings of patients with unilateral disease have a 1% risk when there is no previous diagnosis of retinoblastoma.5
Histopatholopgy of Retinoblastoma
At minimal magnification, more basophilic areas with calcification inside the tumor can be seen in along with immunological areas of necrosis. Not sufficiently differentiated Cells with tumors are round, smaller to average-sized cells with small cytoplasm, large hyperchromatic nuclei, and mitotic data. Tumors that have been well-differentiated demonstrate fleurettes and rosettes. These may appear in a wide range of forms. Columnar cells surrounding a central lumen compose the Flexner Winter Steiner rosettes. This is a highly characteristic of medulla epithelioma as well as retinoblastoma.
The term "pseudo rosette" describes the way tumor cells cluster surrounding blood vessels. These do not suggest effective differentiation. Eosinophilic structures identified as fleurettes are formed up of tumor cells that have pear-shaped eosinophilic processes that increase through a barrier with apertures. The existence of rosettes and fleurettes indicates photoreceptor variations in the tumor cells. Likewise, the inner surfaces of blood vessels' lumen contain deposits known as basophilic deposits, which are precipitated DNA released because of tumor necrosis.4, 6
Diagnosis
Retinoblastoma generally appears between birth and age five, though it can occur later. The typical age for presentations in the US is 18 months (about 1 and a half years). No sex preference is available. Approximately two thirds of cases of retinoblastoma are unilateral, and barely one third are bilateral. The most prevalent presentation includes the pupil's cat's eye reflection. A uveitis-like appearance aseptic orbital cellulitis, phthisis bulbi, hyphemia, buphthalmias, red, painful eye with glaucoma and cloudy cornea, proptosis, and even fungating mass belong to the less common clinical presentations. Patients who have spread may also illustrate headaches, nausea, bone pains, or masses on their scalps. It is likely that less than 10% of cases have family history. The average lifespan at which patients with retinoblastoma provided was 2.5 years in a study done at the All-India Institute of Medical Sciences (the average age for unilaterally retinoblastoma was 3 years, and the median age for bilateral retinoblastoma was 2 years). 29.5% of the cases were bilateral, and 69.9% of the cases were unilateral. With a male to female ratio of 1.6:1, there was a predominance of men. Parents often observed white reflex as the first symptom, then redness or squint. Apart from white reflex, proptosis was the most typical symptom (31.3%) at presentation. The symptoms remained for a median of 7.2 months before their presentation. Thirty percent of the patients experienced intraocular disease, thirty-seven percent had regionally advanced disease, and ten percent had metastatic disease.6
Diagnostic Approach
Numerous benign conditions of the eyes can mimic retinoblastoma. Most can be distinguished with the help of supplementary testing and an appropriate assessment. Coats' Disease is one of the most prevalent illnesses that mimic retinoblastoma. This irregularity in the retinal vasculature has an unknown cause. The patients are marginally older than the 4–10-year-old age range of most children with retinoblastoma. Males are more likely to experience unilateral cases.6
External Examination
Findings like eye strabismus, proptosis, leukocoria (white papillary reflex), or extraocular expansion of a solid object (in advanced cases) should be noted using a penlight. External photos are crucial for medical validation of results. 6
Slit Lamp Examination
Since youngsters (under the age of five) make up most retinoblastoma patients, it might be challenging to do an accurate slit lamp examination in the office due to uncooperative patients. An operating microscope or a portable slit lamp can help you see the anterior segment more clearly after you're in the operating room and the youngster is unconscious. Records should be kept of any findings, including hyphemia or pseudo hypopyon, anterior chamber seeds, anterior chamber invasion by tumor, and neovascularization of the iris.6
Intraocular Pressure Measurement
A Tono pen, Perkins, or Schiotz tonometer should be utilized to measure the intraocular pressure in each eye. Investigations of the corneal diameters are required to identify globe dysmorphism, enlargement, or secondary glaucoma.6
Indirect Opthalmoscopy
The most important phase in the diagnosis of a patient with suspected retinoblastoma is binocular indirect ophthalmoscopy. Initially, it can be done in the office with help from staff members who are holding patients to make the examination easier. Indirect ophthalmoscopy is done in the office with the baby firmly wrapped in a blanket to minimize movement after a topical anesthesia is applied to the eye and an eyelid speculum is placed. While the clinician does a scleral depression examination to document all tumors and related findings, assistants ensure the child's stability and safety. In addition, fundus photography can be done in the office with the infant safely swaddled.6
Under anesthesia, indirect ophthalmoscopy is carried out again at the examination site with a more comprehensive evaluation to record every detail, especially retinal detachment, bleeding, seeding, and every tumor's position, size, and characteristics. At that period, an extensive large-fundus drawing.6
Classification
"The International Classification of Retinoblastoma" is the current classification that is used for organizing and grouping the disease. The older Reese Ellsworth has been replaced by it.
Categorization, since it was not applicable to the methods of treatment used today.
There are five stages
Stage 0: When a child has intraocular retinoblastoma without any systemic or regional tumors, and no enucleation process has been used.
Stage 1: Following one eye's enucleation.
Features associated with high-risk pathology might exist in the enucleated specimen. That could be in the other eye.
Stage 2: After enucleation, when a residual orbital tumor is visible at the optic nerve's cutend.
Stage 3: When there is a noticeable extension of the orbit that involves the cervical or preauricular lymph nodes.
Distant metastasis is stage four. It's separated into:
Stage 4:(a) Spread of the non-central nervous system (CNS), (b) CNS spread in stage 4b.
Assigning each eye group:
There are five groups in this as well
Group A: Tiny tumors less than 3 mm (about 0.12 in) in size that need to be placed 1.5 mm (about 0.06 in) from the optic disc and at least 3 mm (about 0.12 in) from the fovea.
Group B: Any tumor larger than 3 mm (apart from smaller tumors that fall under group A's explanation and are extremely near the fovea and optic disc). Cuff of exudative retinal detachment <1 quadrant or <5 mm (about 0.2 in) from the tumor base is satisfactory.
Group C: Any tumor that has subretinal or vitreous seeds that are less than 3 mm (about 0.12 in) from the tumor surface, or localized tumor dissemination.
Group D: Any tumor containing subretinal or diffuse vitreous seeds
As an illustration:
Testing is done to determine whether cancer cells have spread from the site of the diagnosis to other parts of the body or within the eye in cases of retinoblastoma.
Staging is the procedure used to determine whether cancer has progressed to other parts of the body or within the eye. Whether retinoblastoma is intraocular—only found in the eye—or extraocular—found outside the eye—is determined by the data acquired during the staging process. To plan treatment, it is critical to determine the stage. Cancer is frequently staged using the findings of the tests and procedures used to diagnose the illness.7
Magnetic resonance imaging, or MRI: A technique that creates a sequence of finely detailed images of internal body parts, including the brain, using radio waves, a magnet, and a computer. Another name for this process is nuclear magnetic resonance imaging (NMRI).7
This test's objectives are to confirm the retinoblastoma diagnosis and determine whether the tumors have only progressed to one or both eyes or throughout the brain. This is prevalent in advanced retinoblastoma cases and would necessitate a visit with our pediatric oncologist for additional testing to rule out further diagnosis and more sophisticated therapy. An MRI is typically performed once a year post diagnosis. 8
Bone scan: A bone scan is a method used to determine whether the bone contains fast dividing cells, such as cancer cells. Through a vein, a very tiny amount of radioactive material is injected and enters the bloodstream. A scanner that simultaneously captures an image of the body detects the radioactive material that accumulates in the cancerous bones. Because cancerous bone cells absorb more radioactive material than healthy bone cells do, cancerous bone areas appear brighter in the image. The degree of cerebral cancer can be assessed with CT alone, without the need for brain isotope scanning or electroencephalograms.9, 8
Treatment and therapy
Preserving the patients' lives is the main goal of therapy. The preservation of the damaged eye's globe and preservation of vision are significant but secondary goals of treatment. 10 For patients with retinoblastoma, there are various therapy options. While some treatments are being studied in clinical trials, others are typical (i.e., the treatment that is currently being used). A treatment clinical trial is a type of research study designed to find new treatment options for cancer patients or to help enhance existing ones. A new treatment may become the norm if clinical trials demonstrate that it is superior to the current standard of care. Participating in a clinical study should be taken into consideration because childhood cancer is uncommon. Only patients who haven't started treatment are eligible to participate in some clinical trials.7
Surgery
Enucleation: The preferred course of treatment for unilateral intraocular retinoblastomas of groups E and D is primary enucleation. Cases of retinoblastoma intraocular that have not responded to Enucleation are also used in the treatment of chemotherapy and conservative methods. Furthermore, most patients of overt orbital disease (stage III) can now be treated with enucleation after receiving two to three courses of neoadjuvant chemotherapy.2
Exenteration: It's a very disfiguring procedure. Exenteration in full is no longer necessary. Currently, it is primarily used in cases where primary or recurring orbital illness is unable to reply to neoadjuvant chemotherapy.2
Chemotherapy
To reduce the tumor volume, two cycles of chemo reduction are often administered prior to chemo thermotherapy (CTT). The synergistic effect of hyperthermia, which is quickly administered following the intravenous infusion of chemotherapy, lends weight to the reasoning behind this method of Transpupillary thermotherapy (TTT) administration. The method for treating hyperthermia is the same as for TTT. The timing of treatment is different. 1-2 hours before the TTT, a single dose of the single-agent chemotherapy carboplatin is administered using this approach. For tumors bigger than 7 mm (about 0.28 in), a second TTT session may be used on day 8. It takes an average of three to four cycles of CTT to achieve acceptable tumor control. By the end of the follow-up period, 90% of tumors are under control. Any tumor larger than 6 mm (about 0.24 in) is considered a failure risk factor. This therapy has the advantage of reducing chemotherapy to a single medication, and local control is equivalent to multiagent regimens; nevertheless, scheduling the timing of administration can present challenges.6
Periocular chemotherapy
The primary issue with systemic treatment is vitreous and subretinal seed recurrence. This is thought to be possible due to restricted penetration of Chemotherapeutic drugs are systemically injected into the avascular sub-retinal and vitreous regions.
Higher effective doses at these areas are made possible using local delivery strategies (periocular), which establishes effective treatment while minimizing the systemic negative effects of chemotherapy. Most facilities employ an aqueous solution of carboplatin (same as used for systemic therapy). In the treatment of groups C and D retinoblastoma, periocular carboplatin has been shown to have a promising therapeutic benefit. However, it is associated with serious side effects, such as changes in ocular motility, necrosis of orbital fat, severe pseudopreseptal cellulitis, and ischemic necrosis with atrophy of the optic nerve leading to blindness. The chemotherapeutic agent's quick dispersion is assumed to be the cause of these side effects. Clinical trials are therefore being conducted to evaluate the effects of periocular carboplatin administered in a fibrin sealant, which permits a regulated release of the medication in the periocular space. With a rat model of retinoblastoma, topotecan in fibrin sealant has also been successfully used subconjunctival to treat the disease.2
Intraarterial chemotherapy
Patients with retinoblastoma have recently tried intraarterial injection of the chemotherapeutic medication melphalan. Researchers from Japan have been looking into chemotherapeutic delivered intraretinally. The effectiveness of the intraarterial injections alone, however, cannot be ascertained because many eyes in their studies also received intravitreal melphalan and external beam radiation therapy (EBRT). Good results following direct intraocular melphalan injections have been shown in data from a phase I/II clinical trial recently carried out by Abramson and collaborators. The ocular artery was selectively catheterized, which should have prevented brain toxicity. In all but one of the nine patients who were enrolled in the research, vision stabilized or improved. The stated side-effect profile was mild. However, many patients also had additional brachytherapy, laser, thermotherapy, or EBRT, so it's challenging to say whether this technique alone is beneficial in raising globe salvage rates. While intraarterial chemotherapy has shown exceptional control over retinoblastoma, especially in cases where tumor seeds have returned after prior therapies, several experts caution against its usage. Recently, Shields et al. have evaluated the toxic effects of irradiation from fluoroscopy during intraarterial chemotherapy for retinoblastoma, and have cautioned that accumulated irradiation toxic effects following multiple sessions of intraarterial chemotherapy could be cataractogenic and possibly carcinogenic. 2
Radiation
For the following conditions, radiation therapy in Groups IV and V Rb may be a good alternative to enucleation or a supplement to postoperative care.
When a tumor is treated in one or both eyes, the degree of uninvolved retina is thought to provide a fair probability of vision following tumor irradiation.
After a more badly afflicted eye is removed, a less affected eye is treated in the hopes that some vision will survive.
Treatment for both badly damaged eyes when it is impossible to decide which eye should be treated and which should be removed.
After enucleation, to radiation treat any remaining or recurring orbital tumor
Palliation of a metastatic tumor outside of orbit.
Other local ocular therapies like photocoagulation or cryotherapy can be used in conjunction with radiation therapy. Additionally, it can be used with systemic chemotherapy. The quantity and location of the tumors determine the radiation treatment strategy. Local radiation using a radioactive applicator can be used when the tumor is tiny, involving less than 12 mm (about 0.47 in) of the retina, and when photocoagulation or cryotherapy are not appropriate for treating the lesion. 33 The benefit of this method is that it minimizes the danger of radiation problems while also restricting high-dose radiation to the area of the eye containing the tumor. A scleral applicator impregnated with radioactive cobalt 60 or iodine 125 seeds is selected, encircling the tumor base with at least a 1 mm (about 0.04 in) margin. This plaque is sutured over the appropriate area and left in place to deliver 4,000 rads to the apex of the tumor over several days. 35 The applicator is then removed, and the eye is followed for tumor response. Radioactive applicators cannot be utilized if the tumor involves the macula, or retina near the optic nerve head. Tumors in Groups I, II, and III require a dose of 3500 rads administered in three weeks in three portions per week. Tumors in Groups IV and V require 4500 rads in 4 weeks, using 3 fractions weekly as well.8
Conclusion
To summarize, the trip across the domains of genetic research, retinoblastoma therapy, and diagnostic approaches presents a changing scene with significant progress. The convergence of these fields advances our knowledge and capacity to deal with this illness in its entirety. Despite ongoing difficulties, the field's advancements point to a bright future for retinoblastoma research and clinical application. In the fight against retinoblastoma, the integration of developing therapeutics, improved genetic insights, and cutting-edge diagnostics opens the door to better patient care and inspires optimism for a better future.