Introduction
Topical conveyance is the delivery of medication through the skin layers. The drug used to treat cutaneous disorders and universal disease manifestations that affect the skin such as acne and psoriasis respectively is delivered through this route. Its primary goal is to enclose the therapeutic effects of the medicaments on or within the surface of the skin. Several formulations are seen to be used in the topical route delivery such as foams, sprays, medicated powders, and solution.1
Topical drug delivery system is increasingly becoming more popular and useful in the dermatological field - this is because the penetration of many drug substances is affected by the skin since it acts as a mechanical barrier and delivers medicine both consistently and locally. The affordability and ease of use of the epidermis make it highly popular. Despite its low bioavailability and poor retention, the topical delivery system is subject to extensive research to refine its effectiveness, security and reduce its aftermath. Topical drug products can be categorized based on those applied to produce a local effect or systemic effect.2
For efficient local delivery of these therapeutic agents, topical administration is preferred as it is considered convenient and affordable. Achieving optimal concentration of a specific drug at the required duration is vital for designing a therapeutic system, to achieve this is quite challenging thus creating the attention in the past few years. However, factors like poor retention and low bioavailability create drawbacks in the administration of the drugs hence making the common topical drug delivery systems suffer. To achieve success in the topical delivery of drugs, the defensive barriers should be carefully manipulated, and a soluble drug carrier selected.3
Advantages of topical delivery system
They prevent metabolism because they do not get deactivated by digestive and liver enzymes upon reaching the systemic and portal circulation after gastrointestinal absorption.
The drug can be applied to a large surface area as compared to other routes.
The medication can be applied to a specific area.
Gastrointestinal incompatibilities can be avoided.
Medications with minimal half-life or healing window can be administered through this method.
The effectiveness of a drug can be achieved with low doses by continuous drug supply.
Deep Eutectic Mixtures
Researchers formed a eutectic solution by combining a hydrogen bond donor (HBD) with a hydrogen bond acceptor (HBA). In this case, the HBD includes carbohydrates, alcohol, amines, and carboxylic acids, while HBA entails quaternary salts. 6
DESs differ from ILs in that, they emanate from the eutectic solution that has many cationic and anionic entities from Brønsted or Lewis bases and acids. On the other hand, are made up of a single category of distinct cations and anion species. The application of deep eutectic mixtures differs from those of ILs due to their chemical properties despite having similar physical properties to those of other ILs. DESs have low melting points mainly because of their large, nonsymmetrical ions with low lattice energy. To make a DES, researchers the HBD to a metal salt; and, therefore form quaternary ammonium salts. Notably, charge delocalization originating from combining a donor moiety and halide ion causes a drop in the mixture's melting point. The temperatures of hydrogen bonding differ from that of singular components. 7
Objectives
The main objective of this research was to develop a Topical gel containing deep eutectic mixture containing luliconazole.
To improve the solubility and permeability of Luliconazole by incorporating it into the deep eutectic mixture.
To evaluate the prepared gel and determine if the solubility was increased.
Materials and Methods
Materials
Carbopol 980NF was obtained from (Fisher Scientific India Pvt. Ltd). Choline Bitartrate that was used as a hydrogen bond donor was procured from (Acros Organics). Gallic acid was purchase from (Avarice Laboratories Pvt. Ltd, U.P) and methanol was purchased from (Fisher Scientific India Pvt. Ltd). The Active pharmaceutical ingredient Luliconazole was obtained from (Marksan Pharmaceutical Ltd., Goa). The other chemicals were used as received from the research center (Mohali- Punjab).
This research was conducted at Oniosomes Healthcare Pvt. Ltd, Mohali, Punjab for a period of 6 months from 2 January 2021- 30 June 2021
Methods
The heating method was used for the preparation of deep eutectic mixture. This method involves mixing and heating the components with continuous stirring until a homogeneous mixture was formed. The heating temperature usually ranges between 50o C-100oC. Higher temperatures can lead to degradation of the mixture due to esterification process.
For the preparation of the topical gel the dispersion method was used dispersion method. The drug was dissolved in non-aqueous solvent with preservative. Simultaneously the gelling agent was dispersed in water with stirring at 1200rpm for 30minutes. The drug in solution form was then added to the gelling agent solution with stirring until a gel is formed. 8, 9
Preparation of deep eutectic mixture
For the preparation of Deep Eutectic Solvent Mixture (DESM), choline bitartrate and selected carboxylic acids were mixed at different molar ratios. The mixtures were sealed in vials and heated in an oven at 75℃ until homogenous solutions were formed. Subsequently, these samples were stored at room temperature and only those samples that remained liquid were tested as room-temperature solvents for model poorly soluble drugs. Luliconazole was selected as model drug for solubility experiment due to its poor aqueous solubility. To determine the drug solubility in DESMs, a definite amount of drug was added to a blank DESM solvent followed by vigorous vortexing until the excess solid remained undissolved. The resulting solution containing excess drug was left standing for 24 hours at room temperature to ensure equilibrium was attained. 10
Table 1
Screening of carboxylic acid for deep eutectic solvent mixture of luliconazole
In-vitro drug release study
This study was performed through dialysis membrane using Franz diffusion cell apparatus. The dialysis membrane is previously treated with 20% ethanol solution and soaked overnight. The treated membrane was placed in diffusion cell between the donor and acceptor compartments. About 1g of gel was added to the treated membrane and the receptor compartment of the diffusion cell was filled with 7.4 pH phosphate buffer. The whole assembly was fixed on a magnetic stirrer, and the solution in the receptor compartment was constantly and continuously stirred using magnetic beads at 100 rpm, the temperature was maintained at 37 ±0.50°C.
Samples of 1 ml were withdrawn at time interval of 15, 30, 45, 60, 120, 180, 240, 360, 480, 1080 and 1440 min analyzed for drug by using UV method. The receptor phase was replenished with an equal volume of 7.4 pH buffer at each time of sample withdrawal to maintain the sink conditions. 11
Drug content
The drug content of the prepared gels were conducted by dissolving accurately weighed quantity of gel 1g in 10 ml volumetric flask and volume was made up to 10 ml with solvent. The content was filtered through Whatman paper No. 41. 5 ml of above solution was taken into a 10 ml volumetric flask and volume was made up to mark with solvent. The drug content were estimated spectrophotometrically on a UV-VIS spectrophotometer. 12
Viscosity
Viscosity is the resistance of a fluid to flow. The viscosity of the prepared gel was carried out using a rotational viscometer using T-bar spindle (spindle-L4). Spindle was placed into the gel present in a beaker perpendicularly, such that the spindle end does not touch the lower surface of the beaker. The speed of 50 rpm was maintained for spindle rotation and the values were measured when the gel level was stabilized after 30s. 13
Partition coefficient of the drug
This can be defined as the extent to which a substance is distributed between two liquids, (water-water immiscible) and is usually denoted by log p. The majorly used phases are water and n-octanol (oil phase) in the ratio 1:1. 14
When using water and n-octanol respectively the partition coefficient is given by:
Therefore the partition coefficient is the quotient of two drug concentration in n-octanol and water. It can also be expressed as the logarithmic to base 10.
Shake flask method
The partition coefficient study was performed using shake flask method. 50mg of the drug (Luliconazole) was dissolved in 3 ml of the two solvents (n-octanol: Water) together (1:1) and left to stand for 24 hrs. After 24hrs, the two layers were separated by a separating funnel and centrifuged for 15 mins at 3,000 rpm. The absorbance was taken in UV spectrophotometer at the respective λmax after appropriate dilution. 15
Melting point
Melting point is the temperature at which a substance transition its state from solid to liquid. This temperature is taken in a range at the start, when the solid substances begins to change its form to liquid until it has become fully liquid. The melting point apparatus were used to calculate the melting point of the drug. A few quantities of the drug were inserted in a 10-15 mm long thin walled capillary tube, around 1 mm in diameter inside and closed at one end. The capillary holding the sample was suspended to slowly and uniformly heat the samples, and the thermometer was positioned to verify the temperature. The temperature range over which the sample is observed to melt is taken as the melting point. 16
Solubility studies
Solubility is the ability of a chemical substance either a solid or a liquid (solute) to dissolve in a liquid (solvent) to form a homogeneous solution. Solubility depends on the type of solvent used, temperature and pressure.
50mg of the drug was taken into thoroughly washed test tubes containing 3ml of various solvents (methanol, ethanol, acetone, chloroform, water, PBS pH 7.4) for the solubility analysis and the test tubes were tightly closed. These test tubes were shaken for 24hr at room temperature in a water bath shaker at 15,000 rpm. After 24hr they were centrifuged and then each sample was withdrawal of supernatant. After that, the supernatant were diluted correctly and assessed spectrophotometrically. 17, 18
Spreadability
This is the ability of a gel to spread on a surface/skin when applied. The spreadability of the prepared gels were determined using the following technique: 0.5 g gel was placed within a circle of 1 cm diameter premarked on a glass plate over which a second glass plate was placed. A weight of 500 g was allowed to rest on the upper glass plate for 5 min. The increase in the diameter due to spreading of the gels was noted. 19
Physical appearance
The prepared gel was examined for clarity, colour, homogeneity and the presence of foreign particles. 20
FTIR of Luliconazole and excipients
FT-IR (Fourier Transform Infrared) spectrum of any compound or drug gives information about the functional groups present in a particular compound. FT-IR Spectroscopy was used for structure analysis. The salt (KBr) disc technique was employed. Since the KBr has no absorption within the fundamental region of IR spectrum, only the spectrum of sample is obtained. An FT-IR spectrum of Luliconazole and drug plus excipients mixture was recorded for the determination of drug interaction with excipients. 21
The KBr disc was prepared using 1 mg of luliconazole/excipients plus drug in 100 mg of spectroscopic grade KBr which has been dried using IR lamp. Both KBr and luliconazole was mixed and subjected to hydraulic pressure to make disc. This disc was placed in FT-IR chamber, spectrum was recorded within the 4000 - 400 cm-1 region.
Formulation of topical gel of DESM containing Luliconazole
The optimized formulation (N3) containing 7:3 ratio of Choline Bitatrate and Gallic Acid was found to be effective and was selected as best formulation for the conversion of topical gel formulation. Carbomer 980NF was used to prepare the gel base. In a certain volume of optimized formulation, the Carbomer 980NF was added according to the desired concentration. Gels with different carbopol concentration were prepared to obtain the appropriate gel. The common percentage of carbomer gel making material is 1% - 3%. After complete dispersion, it was placed at room temperature till the carbomer powder completely swelled for 24 hours. 1M of NaOH solution was added drop wise with vigorous stirring to adjust the pH. The developed topical gel was evaluated for various parameters. 22
Drug release kinetic studies
In the present study, raw data obtained from in vitro release studies was analysed, wherein data was fitted to different equations and kinetics model to calculate the percent drug release and release kinetics of luliconazole from topical gel. The kinetic models used were a Zero-order equation, First-order, Higuchi’s model and Korsmeyer-Peppas equation. 23, 24, 25
Zero-order kinetics
Zero-order release would be predicted by the following equation
At = A0 – K0t (1)
Where: At = Drug release at time‘t’, A0 = Initial drug concentration, K0 = Zero order rate constant (hr-1)
When the data is plotted as cumulative percent drug release versus time, if the plot is linear then the data obeys zero-order release kinetics and indicates that the release is time-dependant, with a slope equal to K0.
First order kinetics
First-order release would be predicted by the following equation
Log C = log C0 – Kt/2.303 (2)
Where: C = Amount of drug remained at time‘t’, C0 = Initial amount of drug, K= First-order rate constant (hr-1)
When the data is plotted as log cumulative percent drug remaining versus time yields a straight line, indicating that the release follows first-order kinetics indicating it’s an immediate release. The constant ‘K’ can be obtained by multiplying 2.303 with slope values.
Higuchi’s model
Drug release from the matrix devices by diffusion has been described by following Higuchi’s classical diffusion equation: This model is built on the assumptions that initial drug concentration in the matrix is much higher than drug solubility;
The drug diffusivity is constant, and perfect sink conditions are always attained in the release environment. Higuchi was the first to derive an equation based on Fick Ian diffusion to explain the release of a medication from an insoluble matrix as the square root of a time-dependent phase. Follow the simplified Higuchi equation;
Q = [Dε/τ (2A – εCs)] Cst1/2 (3)
Where: Q= Amount of drug released at time‘t’. D= Diffusion coefficient of the drug in the matrix, A = Total amount of drug in unit volume of matrix, Cs = The solubility of drug in the diffusion medium, ε = Porosity of the matrix, τ = Tortuosity, t = Time (hrs) at which ‘Q’ amount of drug is released.
Equation may be simplified if one may assume that Dε, Cs and A are constant. Then equation becomes:
Q=K.t2 (4)
When the data is plotted according to equation i.e., cumulative drug released versus square root of time, yields a straight line, indicating that the drug was released by diffusion mechanism and it’s a sustained release. The slope is equal to ‘K’.
Korsmeyer-Peppas Model
The release rates from controlled release polymeric matrices can be described by the equation:
Q = K1tn (5)
Where: Q = Percentage of drug released at time‘t’, K = Kinetic constant incorporating structural and geometric characteristics and ‘n’ is the diffusion exponent indicative of the release mechanism.
For Fickian release, n = 0.45 while anomalous (Non-Fickian) transport, n ranges between 0.45 and 0.89 and for zero-order release, n =0.89
Results and Discussion
Organoleptic properties
The organoleptic characteristics of Luliconazole was studied and established. 26
Melting point
The temperature at which a substance transition its state from solid to liquid is known as melting point. Melting point is normally used to signify the purity of the drug. The melting point of luliconazole was studied and recorded. The results are as shown in Table 3 . 27
Solubility studies
The solubility of the drug in different solvents was studied and a solvent to be used in the final formulation was selected. The solubility was analysed using UV-VIS spectrophotometer at 296nm.28
Partition coefficient determination
Partition coefficient is normally used to determine the lipophilic and hydrophilic nature of a substance. Compounds with log p more than 1 are lipophilic in nature whereas those with less than 1 are hydrophilic. 29
Determination of absorption maxima in methanol
UV-VIS spectroscopy is used for quantitative research and is used as a tool for structure elucidation among other uses. The major principle that UV spectrophotometer works by is that when a solution containing the sample is placed in a beam of light, some of the light is reflected, some is absorbed while some is transmitted. A plot of absorbance verses wavelength was made. A double beam UV spectrophotometer in the range 400nm-200nm was used for quantitative elucidation of luliconazole. The outcome is shown in Figure 1.
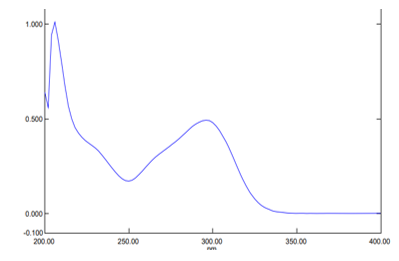
FTIR studies
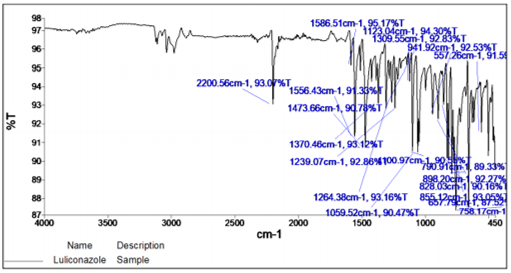
The major peaks were observed at 2200 cm-1 for C≡N stretching, 1586.51 cm-1 C=C alkene stretching, 1473 cm-1 aromatic C=C stretching, 758.17 cm-1 C-Cl bond stretching.
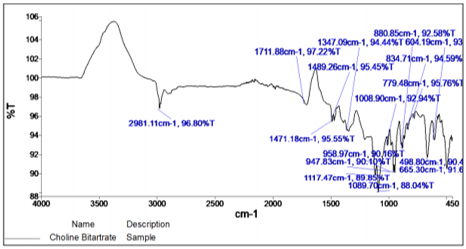
The principle IR absorption peaks of Choline bitartrate were observed at 2981.11 cm-1 (-CH group), 1489.26 cm-1(CH2 bending), 1471.18 cm-1(N-H bond), 1347.09 cm-1(C=O).
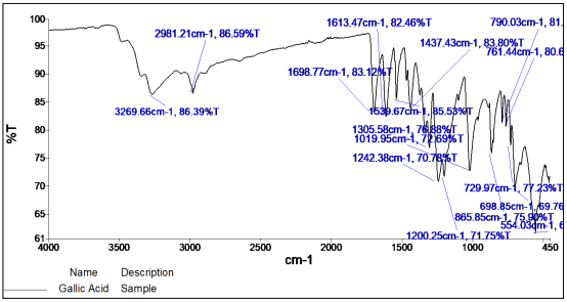
The principle IR absorption peaks of Gallic Acid were observed at 3269.66 cm-1 (O-H stretching), 2981.21 cm-1 (aromatic C-H stretching), 1698.77 cm-1 (aromatic C=O stretching), 1613.47 cm-1 (aromatic C-H stretching), 1539.67 cm-1 (aromatic C-H stretching), 1437.43 cm-1 (aromatic C-H stretching) and 1242.38 cm-1 (phenolic O-H bending). 30
Appearance and pH of Topical gel Luliconazole
The pH and appearance of topical gel of DESM with luliconazole is shown in Table 7.
Viscosity of topical gel luliconazole
The viscosity of topical gel of DESM with luliconazole is shown in Table 8.
Spreadibility of topical gel luliconazole
The spreadibility of topical gel of DESM of luliconazole is shown in Table 9.
Percentage drug content of topical gel luliconazole
The percentage drug content of topical gel of DESM with Luliconazole is shown in Table 10.
Table 10
% Drug content of topical gel luliconazole
|
Formulation Code |
% Drug Content |
|
G1 |
73.45±0.916 |
|
G2 |
78.45±0.916 |
|
G3 |
85.64±0.916 |
|
G4 |
94.24±0.599 |
Formulation G4 was selected for further release study.
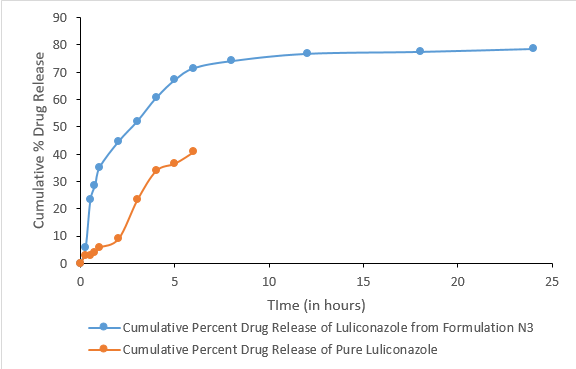
In-vitro drug release study
The in-vitro drug release of Formulation G4 was given in a Table 11.
Table 11
In-vitro drug release study of topical gel of luliconazole
Discussion
The in-vitro release of the drug from the topical gel of luliconazole revealed a significant difference in drug release rate between both the pure drug and DESM. The data of drug retention of 24 hours have shown maximum drug retention (78.58±0.267) of luliconazole from topical gel. Furthermore, the release profile of luliconazole from topical gel showed that it yielded a controlled release profile. From the in-vitro drug release study it was found that pure drug showed lower drug release within short time period. 31
In- vitro release Kinetics
In-vitro drug release kinetic study data of formulation G4 is given below.
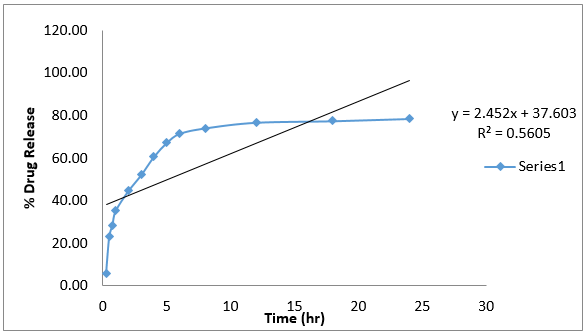
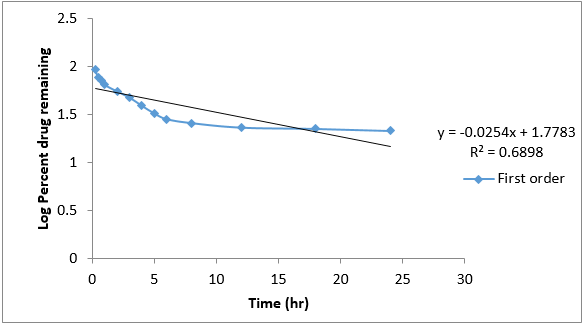
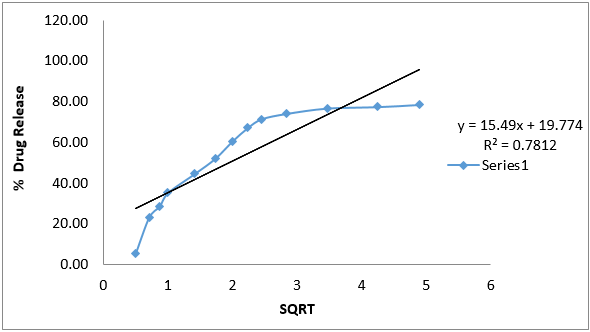
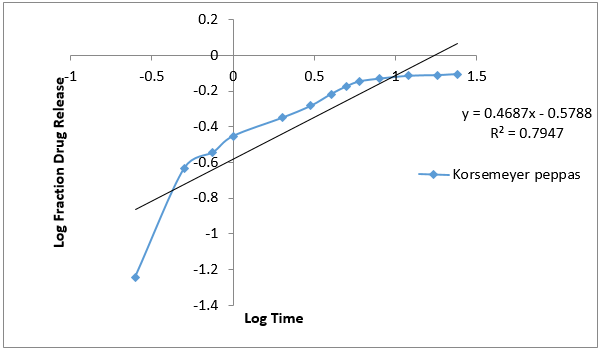
Table 12
Kinetic equation parameter of formulation G4
|
Formulation Code |
Zero order |
First order |
Higuchi |
Peppas |
||||
|
K0 |
R2 |
K0 |
R2 |
K0 |
R2 |
K0 |
R2 |
|
|
G4 |
2.452 |
0.5605 |
-0.0254 |
0.6898 |
15.49 |
0.7812 |
0.4687 |
0.7947 |
The data obtained for in-vitro release shown in Table 10 were fitted into equation for the zero- order, first- order, Higuchi and Korsmeyer- Peppas release models. The interpretation of data was based on the value of the resulting regression coefficients. The calculated regression coefficients for zero- order, first- order and Higuchi models and Korsmeyer-Peppas were shown in Table 11 it was found that the in-vitro drug release of G4 was best explained by Korsemeyer- Peppas model as the plot showed the highest linearity. The value of R2 found to be 0.7947 signifying the highest for the Korsemeyer- Peppas model.
Conclusion
From the data above we can easily conclude that the drug Luliconazole whose permeability and solubility are poor was found to have increased solubility and permeability when incorporated in a deep eutectic mixture thus its safe to say that deep eutectic mixtures can be used to enhance drugs property without interfering with their physiochemical properties.
Conflict of Interest
We authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgments
I want to take this opportunity to say thank you to my Parents, Siblings and a dear friend Jared for their support and for their financial aid, without you this wouldn’t be possible.
Would also like to thank my co-authours for their tireless input
Finally I want to appreciate Oniosomes Healthcare Pvt. Ltd for providing their facilities to conduct this project.