Introduction
By inserting a molecule (ligand) into the chosen binding site of the target specific area of the DNA/protein, For rational drug design and discovery as well as in the mechanistic investigation, molecular docking provides an interesting framework for understanding drug-biomolecular interactions. Primarily non-covalently to create a stable complex with greater specificity and potential efficacy (receptor).1, 2 Molecular docking is one of the most popular virtual screening techniques, especially when the target protein's three-dimensional structure is known. Our method was able to predict the structure of the protein-ligand complex and the binding affinity between the ligand and protein for lead optimization. Molecular docking has been used for more than three decades, and as a result, a large number of novel medications have been found and created.3 Putting molecules in the right configurations so they can interact with a receptor is a process called molecular docking. In the discipline of molecular modeling, docking is a technique that forecasts the ideal positioning of one molecule to another when attached to another to form a rigid complex. By utilizing scoring functions, for instance, it is likely to forecast the ability of suggestion or binding attraction between two molecules using the information on the preferred orientation.
Due to its capacity to anticipate the binding conformation of small molecule ligands to the proper target binding site, molecular docking is one of the most widely employed techniques in structure-based drug design. In addition to helping to clarify underlying biochemical processes, the characterization of binding behavior is crucial for the rational design of pharmaceuticals.
A useful framework for understanding drug biomolecular interactions is molecular docking. Interactions are important for rational drug discovery and design as well as in mechanistic investigation by introducing a chemical (ligand) into the target's chosen binding site mostly non-covalently to form a specific area of the protein (receptor). higher specificity and a stable compound of potential efficacy.4

Application of Molecular Docking
Drug development uses for molecular docking
Docking is very frequently utilized in drug discovery because utmost medicines are made up of small chemical molecules. Using docking, you can:
Hit Identification
To quickly search through a huge data bank of probable drugs in silico for substances that can fix to a certain target of interest, docking in combination with a score function is useful.
Lead Optimization
The binding mode or pose also known as the site and docking can be used to predict the relative position of the interaction between a ligand and a protein. This information can be utilized to create more potent and specific analogs 5
Remediation
Protein-ligand docking can also be used to forecast the substances that enzymes will break down. It can be used to identify the best spot and gather the most effective drug. Enzymes and their modes of action can be found using molecular docking. It is useful for determining the relationships between proteins. The remedy process virtually screens molecules.
Poses vs Binding Location
Binding Site or Active Site
A region of the protein wherever the ligand binds.
On the protein surface, there is typically a cavity.
Can be identified by observing the crystal surface of the protein-bound with a well-known inhibitor.
A binding site is a position on a macromolecule, like a protein, in biochemistry and molecular biology that exactly interacts with another molecule. The binding component of the macromolecule is known by the term "ligand."6
Poses or Binding mode-A common term for describing the geometry of a specific complex is "position" also called "binding mode"7
The binding site's geometry for the ligand.
Geometry is the study of place, orientation, and confirmation.
The process of "docking" molecules into a macromolecular target's binding site involves analyzing their conformation and orientation, which are collectively referred to as their "position." Poses that could be taken are generated by search algorithms and scored by scoring functions.8
Components of Docking Software
Three major parts that function together are often present in Protein-Ligand docking software:
Molecular representation
A method for illustrating structures and qualities (atomic, surface, grid representation) 9
Scoring function
Estimates a score or binding affinity for a certain location.
They give to-
The orientation of the molecules in the binding site.
A score indicating the degree of binding or binding affinity.
Docking Strategies
The idea of molecular complementarity is used in computational docking. The structures fit together like a hand and a glove because of their shape and physicochemical characteristics.
Shape complementarity
The protein and the ligand are seen from one perspective as interdependent surfaces, which employs a matching ability to characterize them.
Simulation
The pairwise interaction energies between ligand and protein are computed in the second perspective by simulating the real Docking process.
Shape complementarity
According to the geometry identical/shape complementarity technique, the protein and ligand have specific established properties that enable docking.
The molecular surface and complementary surface descriptors may be among these characteristics.
The molecular surface of the receptor is defined in terms of its solvent-accessible surface area, whereas the molecular surface of the ligand is described here in terms of its corresponding surface description.10
In this method, the ligand and target are used as superficial structure features that enable molecular interaction (Figures 2 and 3). Here, the exposed surface of the target is represented by the molecular surface of the ligand as equivalent to its solvent-accessible surface area. The identification of the groove for a ligand on the target surface is made easier by the complementarity between two surfaces based on the shape-matching description. For example, the number of shots in the main-chain atoms of protein target molecules is used to determine hydrophobicity. This method is rather fast and involves quickly scanning dozens or even hundreds of ligands in a couple of seconds to discover any potential ligand-target molecular surface binding properties. 11, 12
Figure 2
A shape complementarity strategy is demonstrated in docked adducts. The surface structure characteristic of the ligand and target in this case facilitates their molecular interaction
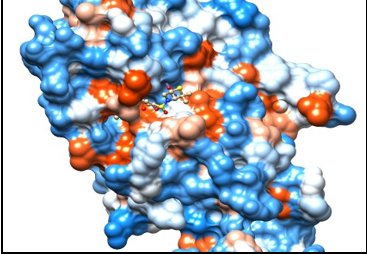
Figure 3
Molecular docking of B-DNA [with sequence (CGCAAATTTCGC)2] dodecamer with anticancer hetero-steroid
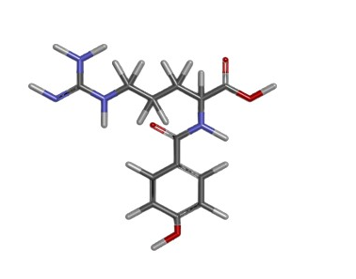
The complimentary pose of docking of the target and ligand molecules can be discovered thanks to the complementarity between the two surfaces with shape-matching descriptions. Another method is to depict the hydrophobic characteristics of the protein by using twists in the main-chain atom. Shape complementarity techniques are typically mountable to even protein-protein interactions and can rapidly scan through thousands of ligands in a matter of instants and evaluate their ability to fix at the protein active site.13
Simulation
The docking process is substantially more difficult to design. Subsequently a certain number of "moves" in the conformational space of the protein, which creates a physical barrier between the protein and ligand, the ligand enters the active site of the protein. Internal structural deviations to the ligand, including torsion angle revolutions as well as rigid body transformations like translation and rotation, are involved in the movements.
The ligand releases energy as "Total Energy" with each movement it makes in the conformational limit. The benefit of this strategy is that it is extra suited to take ligand flexibility. Furthermore, evaluating the molecular credit between the ligand and the target is more accurate.14 This approach takings longer to forecast the ideal docked conformer since each conformation involves a large energy loss. Recently, this problem has been largely revolutionized by the use of quick optimization techniques and grid-based tools to make the simulation approach more user-friendly.15, 16
A docking tool uses this protein structure and a database of potential ligands as involvements. The success of a docking program is subject to two parts:
Docking search in the conformational space
The search space is the set of protein and ligan pairings in all conceivable orientations and confirmations, according to theory. The search space cannot, however, be fully explored in practice using the computational capabilities available today. This required calculating every possible deformation of each particle or molecule because they are energetic and can exist in a variety of conformational conditions, as well as every possible rotation and translation of the ligand concerning the protein.17 stochastic techniques a population of ligands or a ligand conformation at random to explore the conformational space.16
Ligand flexibility
Because it is frequently unclear which shape of a ligand relates most favorably with a receptor, conformational screening is required during docking. Ligand flexibility methods fall into one of three categories: systematic, stochastic, or deterministic searches.
Systematic algorithm
The foundation of systematic search algorithms is a grid-iron of values for each recognized mark of freedom, and every value in the grid is investigated combinatorially throughout the search. The quantity of assessments required rapidly grows with the sum of degrees of freedom. Termination criteria are introduced to address this issue and stop the algorithm from sample spaces that are known to produce incorrect results. Ex. anchor-and-grow or incremental structure algorithm
Stochastic algorithm
The system is altered randomly through stochastic search techniques, usually one degree of freedom at a time. The unpredictability of convergence is one of the main issues with stochastic searches. Numerous separate runs may be carried out to enhance convergence. Ex. Monte Carlo Method.
Deterministic algorithm
The starting state in deterministic searches dictates the change that can be taken to produce the following state, which often takes to have energy that is equal to or lesser than the original state. The same ultimate state will be produced by deterministic searches using the same parameters and the same beginning system. Ex. Energy Minimization Method & Molecular Dynamic Simulation.18
Receptor flexibility
The flexibility of the receptor in docking techniques remains a challenging problem. This challenge is mostly caused by the substantial number of degrees of freedom that must be taken into account during calculations. Neglecting it causes subpar docking outcomes for predicting binding pose. This is a significant challenge when handling flexible proteins for docking. The ligand that a biomolecule or protein binds to determines its conformation. As a result, it is clear that docking with a stiff receptor only produces one receptor shape. However, the ligands may need to bind to numerous receptor conformations if a flexible receptor is being employed for docking. Usually, the protein feature that is neglected the most in molecular docking research is the many protein conformational states. Since p flexibility affects the ability to achieve higher affinity between a given medication and target, it is essential. Target flexibility also includes the water molecules at the active site. Water molecules need to be adjusted if object waters must be prevented during docking. Multiple stationary structures for similar proteins that were experimentally found in various conformities are routinely used to match receptor flexibility. Searching rotamer collections of side chains of amino acids surrounding the binding cavity can result in alternative but energetically sensible protein conformations.19
Scoring function
Scoring functions are precise procedures used to nearly forecast the binding affinity of two molecules afterward they have stayed docked in the fields of computational chemistry and molecular modeling. The two molecules are most frequently a biological target of the medicine, such as a protein receptor, and a small organic substance like a drug.20
Four fundamental classes are used to categorize scoring functions in the docking field:
Force-field
A force field can be used to calculate affinities by adding the electrostatic interactions and intermolecular van der Waals interactions that occur between each atom of the two molecules in the complex. It is also normal to combine the strain energies, or intramolecular energies, of the two binding partners. There are times when implicit solvation methods are used to account for the desolvation energy of the protein and its ligands like GBSA (molecular mechanics generalized Born surface area) or PBSA (molecular mechanics Poisson-Boltzmann surface area). Finally, the presence of water is often required for the binding to occur.21
Empirical method
The empirical method is based on the enumeration of the sum of several kinds of interactions between the 2 binding partners. For a protein-ligand complex with a recognized three-dimensional structure, a scoring function is described that empirically predicts the free energy of binding.22 It is possible to count the ligand and receptor atoms in direct contact or to calculate the SASA difference between the complex and uncomplexed forms of the ligand and protein. It is typical to fit the coefficients of the scoring function using various linear regression techniques. These functionally connected interactions, as example:
Hydrophobic- hydrophobic contacts (favorable)
Unfavorable hydrophobic-hydrophilic interactions (analyses of hydrogen bonds that are not met, a significant enthalpic contributor to binding).23
The number of hydrogen bonds (favorable contribution to affinity, particularly when shielded from solvent; no contribution when the solvent is exposed));
The proportion of connections that are immobile during complex formation but can rotate (an unfavorable contribution to conformational entropy).
Knowledge-based
based on a mathematical analysis of tightly connected intermolecular interactions found in significant 3D databases (like the Protein Data Bank or the Cambridge Structural Database), which is then applied to determine statistical "potentials of mean force". Based on the idea that close contacts between molecules, which happen more frequently than one might expect by chance, are likely to be energetically favorable and hence have a positive impact on binding affinity, this strategy is used.24
Machine learning
The connection between binding affinity and the protein-ligand complex's structural characteristics is not assumed to take a fixed functional shape in machine-learning scoring systems.25 When it comes to determining the binding affinities of various protein-ligand complexes, machine-learning scoring methods have repeatedly been demonstrated to perform better than traditional scoring algorithms.26
The distinction between the first three categories force-field, empirical, and knowledge-based is made based on the assumption that each category's contributions to binding are linearly connected. This limitation prevents standard marking methods from utilizing huge training data sets.27
Disadvantages
There are many X-ray crystallographic structures for systems involving proteins and high-affinity ligands, but there are only a few for systems involving low-affinity ligands since the latter complexes tend to be less stable and hence more challenging to crystallize.
Scoring functions are capable of precisely docking ligands with high affinities, but they can also offer docked conformations for ligands that are unable to attach. This outcomes in a huge number of incorrect positive hits, or ligands that, when combined in a test tube, fail to bind to the protein as predicted.
Recalculating the energy of the highest scoring poses consuming more precise, but computationally additional demanding approaches like General Born or Poisson-Boltzmann methods is one option to lower the amount of false positives.
Imperfections in the scoring function present another difficulty for docking. The primary flaw in molecular docking programming is the lack of a scoring mechanism that is both accurate and quick 28.

Rigid Body Docking
This method of docking maintains the rigidity of both the target and ligand molecules. The rigid body docking program DOCK first establishes how a molecule will be oriented in a binding site before evaluating how well it will fit in that site to fit molecules into receptors. DOCK determines positioning by comparing the internal distances between ligand atoms to the inner distances between sites computed in advance in the receptor binding area.
Flexible Ligand Docking
The target is treated as a stiff molecule in this kind of docking. As seen in Figure 5, this is the docking approach that is most usually employed. 29 Both Target & Ligand are Integrated as Rigid Molecules.
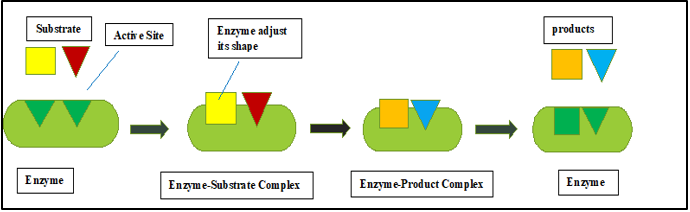
Lock & Key/Rigid Docking
Emil Fischer first proposed the "lock-and-key concept" in 1890, which, as seen in Figure 6, describes how biological systems work. A substrate fits into a macromolecule's dynamic site similarly to how a key fits into a lock. Unique stereochemical characteristics of biological locks are required for their activation.30 The lock and key theory postulate rigidity and tight interaction between ligands and receptors. It defines complementarity’s core Principles in three dimensions.31
Induced Fit/ Flexible Docking
Daniel Koshland first proposed the "induced fit theory" in 1958. The important principle is that during the recognition process, the ligand and target are mutually adaptable by small conformational changes, until In Figure 7, the best fit is made.
Different Types of Interaction in Molecular Docking
The magnitude of the forces among the molecules carried by the elements is whatever is known as the interaction between atoms. These forces are mostly divided into 5 types are given in. Figure 8
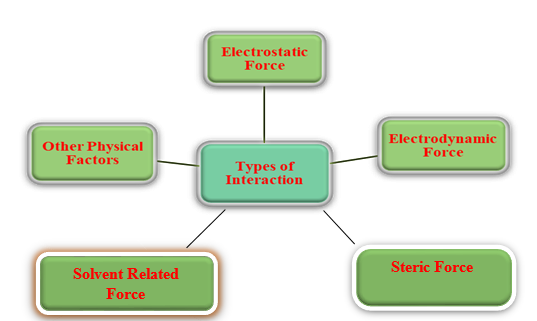
Electrostatic Force: Due to the charges present in the substance, these forces include the dipole-dipole, charge-charge, and charge-dipole interaction forces.
Electrodynamic Force: This involves the distance-dependent Van der Waals interactions between atoms or molecules. At longer distances between two interacting molecules, this disappears.32
Steric Force: Steric forces are non-bonding relations that affect an ion's and molecule's shape and reactivity. Chemical reactions and a system's free energy can be influenced by the forces that result from this.33
Solvent-Related Force: These forces were produced as a result of a chemical reaction between the protein or ligand and the solvent. Examples include hydrogen bonding, which can affect a ligand's or protein's solubility through hydrophilic and hydrophobic interactions.34
Other Physical Factors: The solubility and binding energy of proteins are affected by a wide variety of other forces and interactions.35
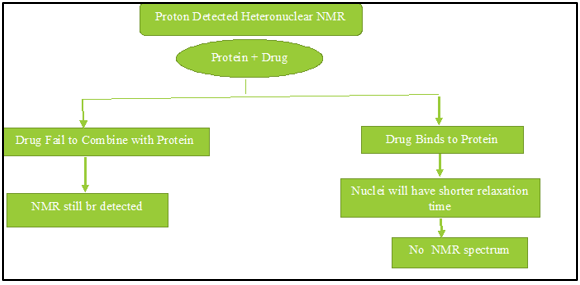
Mechanism of Molecular Docking
Before running a docking screen, the target protein's structure is required. The structure has typically been determined using a physicochemical technique like X-ray crystallography or, less frequently, NMR spectroscopy. This protein structure and a database of ligands are inputs into a docking tool. A docking program's accomplishment is dependent on two elements, such as the search algorithm and the scoring system. Conformational Space Analysis The search space is the set of protein and ligand combinations in all conceivable orientations and conformations. A given level of granularity would need collecting all possible distortions of each molecule as well as all probable rotational and translational directions of the ligand comparative to the protein, which is not possible with the available computing resources. The majority of docking algorithms are currently being used to take flexible ligands into account, and several of them try to represent flexible protein receptors in Figure 9, Figure 10.36, 11
Major Steps in the Molecular Docking Mechanism
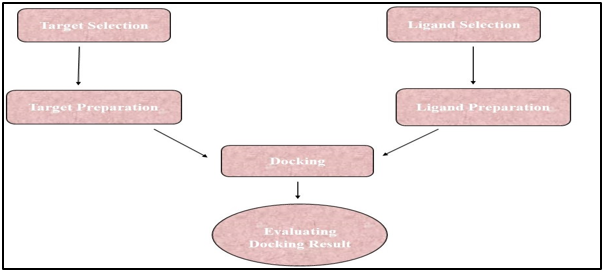
Step I- Protein preparation: A preprocessed 3D structure of the protein would be downloaded from an online database like Protein data bank (PDB). The following adjustments need to be made, as presented in. 40
Step II – Prediction of Active Site: Following the modification and preparation steps for the protein, the active site should be predicted. The target active site should be chosen even though the receptor may have numerous active sites. As seen in the majority of the hetero atoms and water molecules are removed if they are present.41
Step III –Ligand Preparation: The ligand structures can be downloaded from numerous sources, including Pub Chem and ZINC, or they can be sketched using the Chemsketch tool or ChemDraw tool.
The LIPINSKY'S RULE OF FIVE should be applied while choosing the ligand.42 Differentiating between applicants who don't like drugs and those who do can be done using the Lipinski rule of five. For compounds that respond to two or more of the principles, pharmacological similarity assures a high likelihood of success or failure.
To follow LIPINSKY'S RULE of 5, choose a ligand as follows:
No more than five donors for hydrogen bonds
Fewer than ten acceptors of hydrogen bonds
Molecules with masses under 500 Da
High lipophilicity, fourth (expressed as LogP not over 5
Molar refractivity should range from 40 to 130.
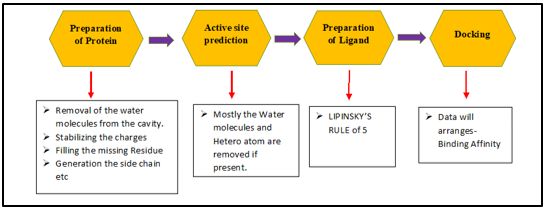
Step IV - The interaction of the ligand with the target protein is investigated. The best-docked ligand complex is used as the basis for the docking software's score and results, and data is analyzed by binding affinity. Numerous docking programs have been developed to perform docking.
Requirement for Molecular Docking
A target protein design, the compounds of interest, or a database containing real or fictitious compounds for the docking process, are the components of a ligand docking strategy. The computational basis enables the application of the proper docking and scoring processes. The ligand is frequently thought of as flexible, while the protein is generally thought of as stiff by docking methods. The location of the protein's bonding within its binding pocket must also be taken into consideration in addition to the degree of structural self-determination. Consensus search, geometric hashing, and pose clustering are some techniques for docking solid molecules or segments onto a protein's active site.
Ligand preparation
To achieve approximated pKa values, hydrogen atoms are frequently added to or removed from the configuration with the maximum probability of becoming dominant.
Receptor preparation
The effectiveness of docking calculations is largely dependent on the quality of the receptor structure used. The observed docking findings will typically be better the higher the employed crystal structure's resolution. A recent analysis of the reliability, constraints, and potential hazards of the structure modification techniques of protein-ligand complexes overall provided a critical evaluation of the existing structures.43
Tools and Software for Molecular Docking Study
Several docking software programs have been developed and made accessible in recent years. The complete description of docking software, which includes the name of the program, the designer or firm, the algorithm, as well as its scoring term and benefits, is described in Table 1.
Table 1
List of docking software tools
Application and Significance of Molecular Docking
As shown in molecular docking analysis is very beneficial for computer-aided drug design (CADD).
Drug development uses for molecular docking: Docking is most commonly used in drug discovery because maximum medicines are made up of small chemical molecules. Using docking, you can:
Hit Identification: When combined with a score function, docking can be used to efficiently search through enormous databases of possible medications for compounds that can bind to a particular target of interest.
Lead Optimization: Docking can be used to forecast the binding mode or posture, commonly referred to as the location and relative position of a ligand's interaction with a protein. This information can be used to create more potent and specific analogs.
Remediation: It is possible to determine which contaminants enzymes will degrade using protein-ligand docking. It can be used to locate the optimal location and gather the most effective drug. Utilizing molecular docking, one can identify enzymes and their mechanisms of action. It can be used to determine how related different proteins are to one another. The remediation process virtually screens molecules.
Conclusion
Molecular docking is a low-cost, safe, and user-friendly technique that assists in the analysis, interpretation, and identification of molecular features utilizing three-dimensional structures. Docking is a technique for anticipating the structural interactions of two or more chemical compounds. The method is utilized in a variety of molecular systems, including material assemblies, big biomolecules, and small molecules., as well as computational chemistry and computer-aided biology. We have briefly discussed molecular docking types, techniques, applications, and challenges in this paper.