Introduction
Inflammation is the protective response of body to external as well as internal untoward stimuli like exposure to harmful substances or internal danger signals released after trauma or cell dysfunction.1, 2, 3 Mankind is always in desire to be healthy and fit. As many diseases such as cancer, metabolic disorders, aging, and neurodegenerative diseases present themselves via inflammation,4, 5, 6, 7 there is always a need to have better anti-inflammatory drugs. The current therapeutic approaches to treat the inflammation includes use of non-steroidal anti-inflammatory drugs and glucocorticoids. These classes of drugs have limitations due to associated severe side effects,8, 9 Thus, there is scope to develop novel effective anti-inflammatory therapeutic agents. Xanthones possesses a unique 9H-Xanthen-9-one scaffold, which occurs mainly in the plants of the families Gentianaceae and Hypericaceae, as well as some fungi and lichens.10, 11 The studies of xanthones are provoking not only due to structural diversity but also due to pharmacological diversity. Xanthone derivatives have shown anti-inflammatory potential12 in previous studies. Thus, we decided to explore the anti-inflammatory potential of novel 3-aminoalkoxy derivatives of xanthone. This research work comprised of theoretically designing thirty 3-aminoalkoxy derivatives of xanthone, estimation of physicochemical properties and their Molecular Docking with COX-1 receptor.
Materials and Methods
Designing and novelty check
Thirty 3-aminoalkoxy derivatives of xanthone were theoretically designed with the general structure- Their novelty was established using Sci-Finder at IIT, Delhi.
Table 1
Physicochemical properties, lipophilicity, water solubility, drug likeness through Lipinski Rule
Table 2
Toxicity profile
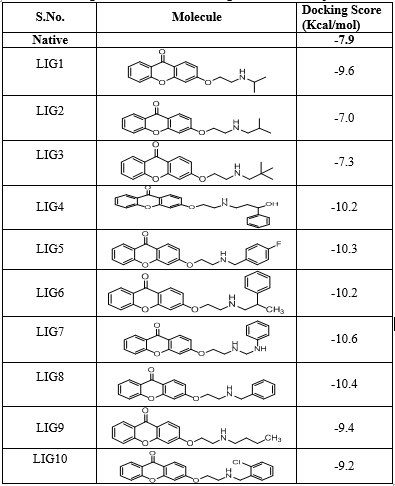
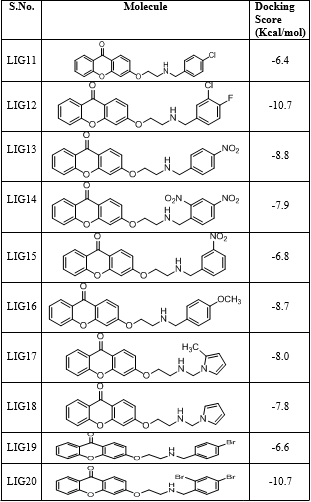
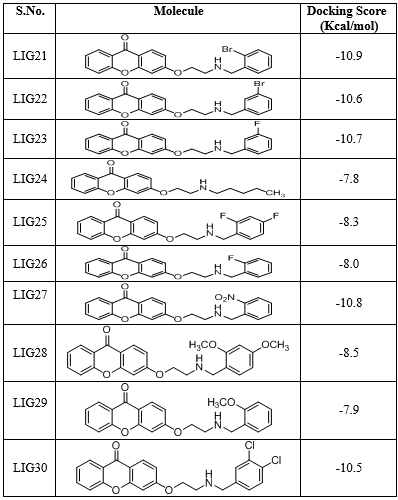
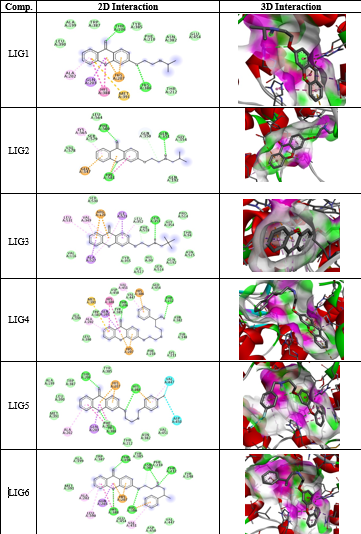
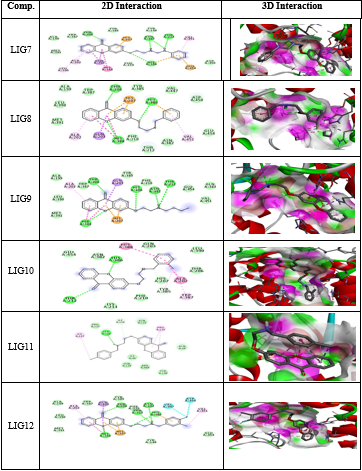
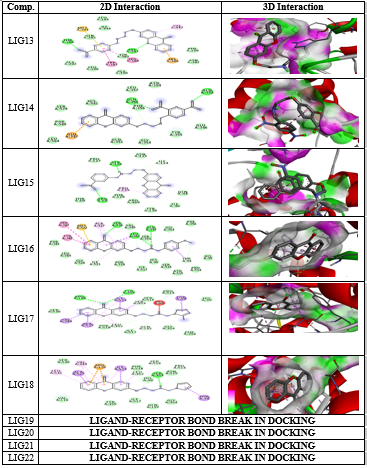
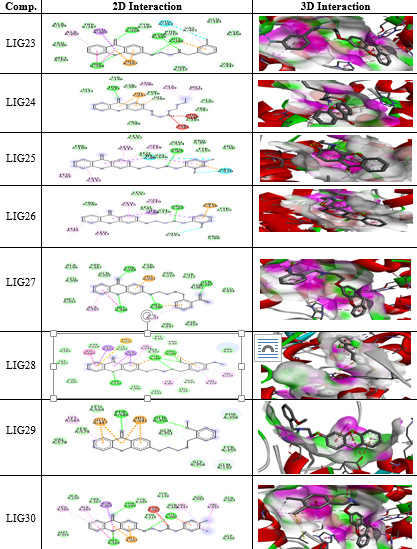
Table 3
Bioactivity score
Estimation of physicochemical properties
Estimation of Physicochemical Properties helps in determining the potential of the designed compounds to become useful drugs by taking into consideration their Absorption, Distribution, Metabolism and Excretion (ADME) drug score and Toxicity profile. The process is relatively fast and inexpensive and can be readily done at the initial stages for the alternatives assessment.13
ADME and toxicity analysis were performed using Osiris Property Explorer, Swiss ADME and Molinspiration softwares.
Molecular docking
The preparation of ligands and receptors were done by the software UCSF Chimera 1.13.1rc (build 41965) developed by University of California. Docking was done by Autodock 1.5.6 developed by molecular graphics laboratory, The Scripps Research Institute, NBCR. Interaction of ligands with receptor was accessed by using Biovia Discovery Studio Visualizer v 17.2.0.16349 developed by Dassault Systemes Biovia Corp.
Results and Discussion
Estimation of physicochemical properties
ADME analysis of the designed compounds include:
Assessing compliance with Lipinski rule and the possible violations: According to the Lipinski rule-of-five, poor absorption or permeation is more likely when the molecular weight is greater than 500, there are more than 5 H-bond donors, 10 H-bond acceptors, and the calculated Log P (CLog P) is greater than 5.14, 15 For good absorption, TPSA value should be between 20-130 Å.2 For good solubility, LogS should not be higher than 6. Molecule become flexible when it does not contain more than 9 rotatable bonds. The designed compound with higher value of drug score and drug likeness is considered as better molecule. As shown in Table 1, all the designed xanthone derivatives, LIG1- LIG30 have adequate number of hydrogen bond acceptors, hydrogen bond donors and rotatable bonds. Solubility, TPSA and cLogP are also in the specified range. Molecular weight of LIG20 is 503.18 which exceeds 500 and hence, a violation of Lipinski Rule of Five. LIG1, LIG4 and LIG17 showed highest values of 2.83, 3.26 and 2.48 respectively for drug likeness. LIG1, LIG5 and LIG18 showed best drug scores of 0.36, 0.4 and 0.37 respectively out of 30 designed compounds.
Assessing Toxicity profile: It includes study of compounds for mutagenicity, tumorigenicity, irritancy and reproductive effects. As shown in Table 2, all the designed xanthone derivatives were found to be mutagenic but none was tumorigenic. No compound showed reproductive system related effects. Except LIG9, all other compounds were nonirritant.
Assessing Bioactivity score: When the bioactivity score is more than 0, the complex is active; if it is in between −5 to 0, the complex is moderately active and if it is less than −5, complex is inactive. Bioactivity score of the designed compounds against various receptors is shown in Table 3. Native ligand Nimuselide showed bioactivity score of -0.15, -0.01, -0.15, -0.17, -0.11 and -0.12 against G-protein coupled receptors (GPCR), Ion channel modulator receptors, Kinase inhibitor receptor, Nuclear receptor, Protease inhibitor receptor and Enzyme inhibitor receptor respectively. LIG4 and LIG24 showed best bioactivity score of 0.21 and 0.04 against GPCR. LIG4, LIG6 and LIG24 showed best bioactivity score of -0.05, -0.14 and -0.17 against ion channel receptor. LIG3, LIG12 and LIG18 showed best bioactivity score of 0.01, 0.03 and 0.03 against kinase inhibitor receptor. LIG4, LIG9 and LIG24 showed best bioactivity score of 0.29, 0.11 and 0.12 against nuclear receptor. LIG4, LIG3 and LIG7 showed best bioactivity score of 0.01, -0.04 and -0.03 against protease inhibitor receptor and LIG4, LIG9 and LIG24 showed best bioactivity score of 0.20, 0.10 and 0.10 against enzyme inhibitor receptor. It was observed that LIG4 showed good bioactivity score against all the six receptors.
M.W.: Molecular Weight; HBA: Hydrogen Bond Acceptor; HBD: Hydrogen Bond Donor; R.B.: Rotatable Bonds; Sol: Aqueous Solubility; TPSA: Topological Polar Surface Area; DS: Drug Score
Molecular docking
All the thirty molecules (LIG1- LIG30) were docked against the 3D structures of COX-1 (Cyclooxygenase-1) receptor (PDB: 3N8X). The Docking Scores are shown in Figure 3 while Figure 7 gives images of 2D and 3D interaction of Ligands (LIG1- LIG30) with COX-1 receptor. The docking scores of LIG21 and LIG27 were found to be -10.9 and -10.8 kcal/mol. It is observed that best docking scores are given by various bromo phenyl substituted derivatives namely LIG20, LIG21 and LIG22 but in docking studies, ligand- receptor bond breaking is shown. Thus, these derivatives are not suitable as drug candidates.
The halo substituted phenyl derivatives such as LIG12, LIG23 and LIG30 showed good docking scores indicating that the electron releasing substitution increases the COX-1 binding ability with the compound. LIG7 has anilino substitution and probably the amino group of aniline is responsible for better binding to the receptor due to its strong electron releasing action. A contradictory result was observed in case of ortho-nitro phenyl substituted derivatives as in LIG27 which shows good binding ability but all the other nitro derivatives exhibited poor binding ability.
Conclusion
Majority of the ssynthesized compounds showed better docking scores than the native ligand, Nimuselide. The halo substituted phenyl derivatives such as LIG12, LIG23 and LIG30 showed good docking scores indicating that the electron releasing substitution increases the COX-1 binding ability with the compound. LIG7 has anilino substitution and probably the amino group of aniline is responsible for better binding to the receptor due to its strong electron releasing action.